
1. Bezeichnung des Arzneimittels
Fibryga 1 g. Pulver und Lösungsmittel zur Herstellung einer Injektions-/Infusionslösung.
2. Qualitative und quantitative Zusammensetzung
Humanes Fibrinogen Jede Flasche Fibryga enthält 1 g humanes Fibrinogen. Nach Rekonstitution mit 50 mL Wasser für Injektionszwecke enthält Fibryga ca. 20 mg/mL humanes Fibrinogen. Die Menge an gerinnungsfähigem Protein wird entsprechend der Monografie des Europäischen Arzneibuchs für humanes Fibrinogen bestimmt. Hergestellt aus dem Plasma menschlicher Spender. Sonstige Bestandteile mit bekannter Wirkung: bis zu 132 mg (5,8 mmol) Natrium pro Flasche. Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
3. Darreichungsform
Pulver und Lösungsmittel zur Herstellung einer Injektions-/Infusionslösung. Das Pulver ist weiß oder blassgelb, hygroskopisch und von fein- bis grobkörniger Konsistenz. Das Lösungsmittel ist eine klare und farblose Flüssigkeit.
4. Klinische Angaben
4.1 Anwendungsgebiete
Behandlung und perioperative Prophylaxe von Blutungen bei Patienten mit kongenitaler Hypo- oder Afibrinogenämie mit Blutungsneigung. Als Komplementärtherapie bei der Behandlung von unkontrollierten schwerwiegenden Blutungen bei Patienten mit erworbener Hypofibrinogenämie im Verlauf eines chirurgischen Eingriffs.
4.2 Dosierung und Art der Anwendung
Die Behandlung sollte unter der Überwachung eines Arztes eingeleitet werden, der über Erfahrung in der Behandlung von Blutgerinnungsstörungen verfügt. Dosierung Dosierung und Dauer der Substitutionstherapie hängen von der Schwere der Erkrankung, der Lokalisation und dem Umfang der Blutung sowie vom klinischen Zustand des Patienten ab. Die individuelle Dosierung sollte auf Grundlage des (funktionalen) Fibrinogenspiegels berechnet werden. Menge und Häufigkeit der Anwendung sollten individuell für den einzelnen Patienten durch regelmäßige Messung des Fibrinogenspiegels im Plasma, durch kontinuierliche Überwachung des klinischen Zustands des Patienten und durch Überwachung anderer gleichzeitig angewandter Substitutionstherapien bestimmt werden. Bei größeren chirurgischen Eingriffen ist eine präzise Überwachung der Substitutionstherapie durch Gerinnungstests unbedingt erforderlich.
1. Prophylaxe bei Patienten mit kongenitaler Hypo- oder Afibrinogenämie und bekannter Blutungsneigung.
Um übermäßige Blutungen bei chirurgischen Eingriffen zu verhindern, wird eine prophylaktische Behandlung empfohlen, um den Fibrinogenspiegel auf 1 g/L anzuheben und auf diesem Wert zu halten, bis die Hämostase unter Kontrolle ist, und auf über 0,5 g/L zu halten, bis die Wundheilung abgeschlossen ist. Für einen chirurgischen Eingriff oder zur Behandlung einer Blutungsepisode sollte die Dosis wie folgt berechnet werden.
Dosis (mg/kg Körpergewicht) =
(Zielspiegel [g/L] – gemessener Spiegel [g/L])
0,018 (g/L pro mg/kg Körpergewicht)
Die nachfolgende Dosierung (Injektionsdosen und -häufigkeit) sollte auf Grundlage des klinischen Zustands des Patienten und der Laborergebnisse erfolgen. Die biologische Halbwertszeit von Fibrinogen beträgt 3 – 4 Tage. Daher ist bei fehlendem Verbrauch eine wiederholte Behandlung mit humanem Fibrinogen in der Regel nicht erforderlich. Aufgrund der Akkumulation bei wiederholter Verabreichung zur Prophylaxe sollten die Dosis und Häufigkeit der Gabe auf Grundlage der therapeutischen Ziele des Arztes für den jeweiligen Patienten bestimmt werden.
Dosierung bei besonderen Patientengruppen
Kinder und Jugendliche
Zurzeit vorliegende Daten werden in den Abschnitten 4.8 und 5.1 beschrieben; eine Dosierungsempfehlung für Kinder kann jedoch nicht gegeben werden.
Ältere Patienten
In klinischen Studien mit Fibryga waren keine Patienten ab 65 Jahren eingeschlossen, so dass keine endgültige Aussage darüber gemacht werden kann, ob diese Patienten anders auf die Behandlung ansprechen als jüngere Patienten.
2. Behandlung von Blutungen
Blutungen bei Patienten mit kongenitaler Hypo- oder Afibrinogenämie
Bei der Behandlung von Blutungen sollte der Fibrinogenspiegel im Plasma auf den empfohlenen Zielwert von 1 g/L angehoben werden. Der Fibrinogenspiegel sollte auf diesem Wert gehalten werden, bis die Hämostase unter Kontrolle ist.
Blutungen bei Patienten mit erworbenem Fibrinogenmangel
Erwachsene
Im Allgemeinen werden initial 1 – 2 g verabreicht mit nachfolgenden Infusionen nach Bedarf. Bei schwerwiegenden Blutungen z. B. bei größeren Operationen, können größere Mengen an Fibrinogen (4 – 8 g) erforderlich sein.
Kinder
Die Dosierung sollte nach dem Körpergewicht und der klinischen Notwendigkeit bestimmt werden, beträgt aber normalerweise 20 – 30 mg/kg.
Art der Anwendung
Intravenöse Infusion oder Injektion. Fibryga sollte bei Patienten mit kongenitaler Hypo- oder Afibrinogenämie langsam intravenös mit einer empfohlenen maximalen Infusionsgeschwindigkeit von 5 mL/min verabreicht werden, bei Patienten mit erworbenem Fibrinogenmangel mit einer empfohlenen maximalen Infusionsgeschwindigkeit von 10 mL/min. Hinweise zur Rekonstitution des Arzneimittels vor der Anwendung, siehe Abschnitt 6.6.
4.3 Gegenanzeigen
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
4.4 Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung
Thromboembolie
Es besteht ein Thromboserisiko, wenn Patienten mit angeborenem oder erworbenem Fibrinogenmangel mit humanem Fibrinogen behandelt werden, insbesondere bei hohen Dosen oder wiederholter Gabe. Patienten, die humanes Fibrinogen erhalten, sollten engmaschig auf Anzeichen oder Symptome einer Thrombose überwacht werden.
Bei Patienten mit koronarer Herzkrankheit oder Myokardinfarkt in der Anamnese, Patienten mit Lebererkrankung, peri- oder postoperativen Patienten, Neugeborenen oder Patienten mit einem Risiko thromboembolischer Ereignisse oder disseminierter intravaskulärer Gerinnung muss der potenzielle Nutzen einer Behandlung mit Fibrinogen aus humanem Blutplasma gegen das Risiko thromboembolischer Komplikationen abgewogen werden. Vorsicht und eine engmaschige Überwachung sind geboten.
Erworbene Hypofibrinogenämie ist mit niedrigen Plasmakonzentrationen aller Gerinnungsfaktoren (nicht nur Fibrinogen) und Gerinnungshemmern assoziiert. Daher sollte eine Behandlung mit Blutprodukten, die Gerinnungsfaktoren enthalten, erwogen werden. Eine sorgfältige Überwachung des Gerinnungssystems ist notwendig.
Allergische oder anaphylaktoide Reaktionen
Wenn allergische oder anaphylaktoide Reaktionen auftreten, muss die Injektion/Infusion sofort gestoppt werden. Im Falle eines anaphylaktischen Schocks muss die standardmäßige medizinische Schockbehandlung durchgeführt werden.
Natriumgehalt
Fibryga enthält bis zu 132 mg (5,8 mmol) Natrium pro Flasche, entsprechend 6,6 % der von der WHO für einen Erwachsenen empfohlenen maximalen Tageszufuhr von 2 g Natrium. Dies ist bei Patienten zu berücksichtigen, die eine kontrollierte natriumarme Diät einhalten müssen.
Virussicherheit
Standardmaßnahmen zur Vorbeugung von Infektionen, die sich durch den Einsatz von Arzneimitteln ergeben, die aus menschlichem Blut oder Blutplasma hergestellt sind, schließen die Auswahl der Spender und das Screening der einzelnen Spenden und Plasmapools auf spezifische Infektionsmarker sowie effektive Schritte zur Inaktivierung/ Entfernung von Viren im Herstellungsverfahren ein. Dennoch kann bei der Verabreichung von Arzneimitteln, die aus menschlichem Blut oder Blutplasma hergestellt wurden, die Möglichkeit der Übertragung von Krankheitserregern nicht völlig ausgeschlossen werden. Dies gilt auch für bislang unbekannte oder neu aufgetretene Viren und andere Pathogene.
Die getroffenen Maßnahmen werden als wirksam angesehen für umhüllte Viren wie z. B. das humane Imundefizienzvirus (HIV), das Hepatitis B-Virus (HBV) und das Hepatitis C-Virus (HCV) sowie für das nicht umhüllte Hepatitis A-Virus (HAV). Die getroffenen Maßnahmen sind bei nicht umhüllten Viren wie dem Parvovirus B19 möglicherweise von begrenztem Wert. Parvovirus- B19-Infektionen können für schwangere Frauen (fetale Infektion) und für Personen mit Immunschwäche oder verstärkter Erythropoese (z. B. hämolytische Anämie) schwerwiegende Folgen haben.
Bei Patienten, die regelmäßig bzw. wiederholt mit aus humanem Plasma gewonnenen Arzneimitteln behandelt werden, sollte eine entsprechende Impfung (Hepatitis A und B) erwogen werden. Es wird auf die Dokumentationspflicht gemäß Transfusionsgesetz hingewiesen.
Immunogenität
Bei Substitutionstherapien mit Gerinnungsfaktoren bei anderen angeborenen Mangelkrankheiten wurden Antikörperreaktionen beobachtet. Für Fibrinogenkonzentrate liegen aber derzeit keine Daten vor.
4.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Es sind keine Wechselwirkungen zwischen Produkten mit humanem Fibrinogen und anderen Arzneimitteln bekannt.
4.6 Fertilität, Schwangerschaft und Stillzeit
Schwangerschaft
Tierexperimentelle Studien zur Reproduktion wurden mit Fibryga nicht durchgeführt (siehe Abschnitt 5.3). Da der Wirkstoff menschlichen Ursprungs ist, wird er in der gleichen Weise wie das körpereigene Protein des Patienten abgebaut. Es ist nicht zu erwarten, dass diese physiologischen Bestandteile des menschlichen Blutes zu Reproduktionsschäden oder zu Schäden bei dem Fötus führen. Die Sicherheit von Fibryga bei Anwendung während der Schwangerschaft wurde nicht in kontrollierten klinischen Studien untersucht. Die klinische Erfahrung mit Fibrinogenprodukten bei der Behandlung geburtshilflicher Komplikationen lässt keine gesundheitsschädlichen Auswirkungen während der Schwangerschaft oder auf die Gesundheit des Fötus oder Neugeborenen erwarten.
Stillzeit
Es ist nicht bekannt, ob Fibryga in die Muttermilch übergeht. Die Anwendung von Fibryga bei stillenden Frauen wurde in klinischen Studien nicht untersucht.
Fertilität
Es liegen keine Daten zur Fertilität vor.
4.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Fibryga hat keinen Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
4.8 Nebenwirkungen
Zusammenfassung des Sicherheitsprofils
Es liegen keine belastbaren Daten zu den Häufigkeiten von Nebenwirkungen in klinischen Studien mit diesem Arzneimittel vor. In klinischen Studien wurden die folgenden Nebenwirkungen berichtet: leichte Pyrexie, die von einem Patienten berichtet wurde, sowie Arzneimittelexanthem, das sich als leichte Hautreaktion mit Juckreiz und Rötung nach Anwendung des Arzneimittels äußerte und ebenfalls von einem Patienten berichtet wurde.
Hinweise zur Sicherheit hinsichtlich übertragbarer Erreger, siehe Abschnitt 4.4.
Die folgenden Nebenwirkungen wurden von Fibryga und anderen Fibrinogenkonzentraten berichtet:
| Standardsystemorganklasse gemäß MedDRA | Nebenwirkungen | Häufigkeit |
|---|---|---|
| Erkrankungen des Immunsystems | Allergische oder anaphylaktoide Reaktionen Hautreaktionen | Nicht bekannt |
| Gefäßerkrankungen | Thromboembolische Episoden (einschließlich Myokardinfarkt und Lungenembolie) (siehe Abschnitt 4.4) Thrombophlebitis | Nicht bekannt |
| Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Anstieg der Körpertemperatur (Pyrexie) | Nicht bekannt |
Kinder und Jugendliche:
Es wurden acht Patienten im Alter von 12 bis 18 Jahren in die Sicherheitsanalyse bei kongenitalem Fibrinogenmangel eingeschlossen.
Das allgemeine Sicherheitsprofil ist bei Erwachsenen und Jugendlichen gleich. Es liegen keine Daten zur Anwendung von Fibryga bei pädiatrischen Patienten mit erworbenem Fibrinogenmangel vor.
Meldung des Verdachts auf Nebenwirkungen
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Straße 51-59, 63225 Langen, Telefon: +49 6103 77 0, Telefax: +49 6103 77 1234, Website: www.pei.de anzuzeigen.
4.9 Überdosierung
Zur Vermeidung von Überdosierungen sind während der Therapie regelmäßige Kontrollen des Fibrinogenspiegels im Plasma angezeigt (siehe 4.2).
Im Falle einer Überdosierung besteht ein erhöhtes Risiko für thromboembolische Komplikationen.
5. Pharmakologische Eigenschaften
5.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Antihämorrhagika, humanes Fibrinogen, ATC-Code: B02BB01 Humanes Fibrinogen (Gerinnungsfaktor I) wird in Anwesenheit von Thrombin, aktiviertem Gerinnungsfaktor XIII (FXIIIa) und Calciumionen in ein stabiles und elastisches dreidimensionales Fibrinnetz umgewandelt. Die Gabe humanen Fibrinogens erhöht den Fibrinogenspiegel im Plasma und kann den Blutgerinnungsdefekt bei Patienten mit Fibrinogenmangel vorübergehend korrigieren.
In einer offenen, prospektiven, randomisierten, kontrollierten, zweiarmigen pharmakokinetischen Cross-Over-Einzeldosisstudie der Phase II bei 22 Patienten mit kongenitalem Fibrinogenmangel (Afibrinogenämie) (siehe Abschnitt 5.2) wurde außerdem die maximale Gerinnselfestigkeit (Maximum Clot Firmness, MCF) als Surrogatmarker für die hämostatische Wirksamkeit bestimmt (FORMA-01). Die MCF wurde mittels Thrombelastometrie (ROTEM) bestimmt. Für jeden Patienten wurde der MCF-Wert vor (Ausgangswert) und eine Stunde nach Gabe einer Einzeldosis Fibryga bestimmt. Die MCF-Werte waren nach der Gabe von Fibryga im Vergleich zum Ausgangszeitpunkt signifikant höher.
Tabelle 1: Maximale Gerinnselfestigkeit (MCF, in mm) (ITT-Population) n = 22
| Zeitpunkt | Mittelwert ± SD | Median (Bereich) |
|---|---|---|
| Vor der Infusion | 0 ± 0 | 0 (0 – 0) |
| 1 Stunde nach der Infusion | 9,7 ± 3,0 | 10,0 (4,0 – 16,0) |
| Mittlere Veränderung (Primäranalyse)* | 9,7 ± 3,0 | 10,0 (4,0 – 16,0) |
MCF = Maximale Gerinnselfestigkeit (Maximum Clot Firmness); ITT = Intention-to-treat.
* p < 0,0001 (95%-Konfidenzintervall 8,37; 10,99)
In einer laufenden prospektiven, offenen, nicht kontrollierten, multizentrischen Studie der Phase III (FORMA-02) wurden 13 Patienten mit kongenitalem Fibrinogenmangel (Afibrinogenämie und Hypofibrinogenämie) in einem Alter von 13 bis 53 Jahren (2 Jugendliche und 11 Erwachsene) einer Zwischenanalyse unterzogen. Diese umfasste die Behandlung von 23 Blutungsepisoden und 4 operativen Eingriffen. Der mittels ROTEM und der Fibrinogenspiegel im Plasma bestimmte MCF-Wert war gegenüber dem Ausgangswert signifikant verschieden. Alle untersuchten Behandlungen bei Blutungsepisoden und operativen Eingriffen wurden vom Prüfarzt und einem unabhängigen Bewertungsgremium unter Verwendung eines objektiven Wertungssystems als erfolgreich eingestuft (Wertung als gute oder ausgezeichnete Wirksamkeit).
Die prospektive, randomisierte, kontrollierte Studie FORMA-05 untersuchte die hämostatische Wirksamkeit und Sicherheit von Fibryga im Vergleich zu Kryopräzipitat als Fibrinogenersatzquelle bei Patienten, die während einer zytoreduktiven Operation zur Behandlung eines extensiven abdominalen malignen Pseudomyxoma Peritonei einen erworbenen Fibrinogenmangel entwickelten. Die Studie schloss 43 erwachsene Patienten in das Per-Protocol (PP)-Analyse- Set ein, 21 mit Fibryga behandelte Patienten und 22 mit Kryopräzipitat behandelte Patienten. Intraoperative Fibrinogensupplementierung wurde präventiv durchgeführt (d. h. 60 – 90 Minuten nach Operationsbeginn, wenn ein übermäßiger Blutverlust beobachtet wurde, aber bevor ein Blutverlust von 2 Litern auftrat) mit Dosen von 4 g Fibryga oder 2 Pools von 5 Einheiten Kryopräzipitat, bei Bedarf wiederholt. Während der 7,8 ± 1,7 Operationsstunden wurden 6,5 ± 3 g Fibryga (89 ± 39 mg/kg Körpergewicht) bzw. 4,1 ± 2,2 Pools von 5 Einheiten Kryopräzipitat verwendet. Im Median wurden intraoperativ 1 Einheit bzw. 0,5 Einheiten EK an Patienten verabreicht, die mit Fibryga bzw. Kryopräzipitat behandelt wurden, bei einem Median von 0 Einheiten EK während der ersten 24 postoperativen Stunden in beiden Gruppen (siehe Tabelle 2). Während der Studie wurde kein gefrorenes Frischplasma (GFP) oder Thrombozytenkonzentrat transfundiert. Die auf Fibrinogensupplementierung basierende hämostatische Therapie wurde von einem unabhängigen Bewertungsgremium anhand eines objektiven Wertungssystems für beide Gruppen als zu 100 % erfolgreich eingestuft.
Tabelle 2: EK* Transfusions-Einheiten intraoperativ und während der ersten postoperativen
24 Stunden (PP-Population)
| Zeitrahmen | Fibryga -Gruppe (n = 21) Median (Bereich) | Kryopräzipitat-Gruppe (n = 22) Median (Bereich) |
|---|---|---|
| Intraoperativ | 1 (0 – 4) | 0,5 (0 – 5) |
| Erste 24 Stunden nach der Operation | 0 (0 – 2) | 0 (0 – 2) |
EK = Erythrozyten-Konzentrat; PP = pro Protokoll.
* Es wurde keine Transfusion mit anderen allogenen Blutprodukten, wie z. B. gefrorenes Frischplasma oder Thrombozytenkonzentrat durchgeführt.
Fibryga wurde in zwei klinischen Studien 8 Patienten im Alter zwischen 12 und 18 Jahren verabreicht. Die Europäische Arzneimittel-Agentur hat für Fibryga eine Zurückstellung von der Verpflichtung zur Vorlage von Ergebnissen zu Studien bei Patienten unter 12 Jahren zur Behandlung eines kongenitalen Fibrinogenmangels gewährt (siehe Abschnitt 4.2 bzgl. Informationen zur Anwendung bei Kindern und Jugendlichen).
5.2 Pharmakokinetische Eigenschaften
Humanes Fibrinogen ist ein normaler Bestandteil des menschlichen Plasmas und verhält sich wie körpereigenes Fibrinogen. Die biologische Halbwertszeit von Fibrinogen im Plasma beträgt 3 – 4 Tage. Fibryga wird intravenös verabreicht und ist sofort in einer der verabreichten Dosis entsprechenden Plasmakonzentration verfügbar.
In einer offenen, prospektiven, randomisierten, kontrollierten, zweiarmigen Cross- Over-Studie der Phase II bei 22 Patienten mit kongenitalem Fibrinogenmangel (Afibrinogenämie) im Alter zwischen 12 und 53 Jahren (6 Jugendliche, 16 Erwachsene) wurden die pharmakokinetischen Eigenschaften von Fibryga nach Gabe einer Einzeldosis mit denen eines anderen auf dem Markt befindlichen Fibrinogenkonzentrats bei den gleichen Patienten verglichen (FORMA-01). Jeder Patient erhielt eine intravenöse Einzeldosis von 70 mg/kg Fibryga und dem Vergleichspräparat. Zur Bestimmung der Fibrinogenaktivität wurden vor Beginn der Infusion und bis 14 Tage nach Ende der Infusion Blutproben gesammelt. Die pharmakokinetischen Parameter von Fibryga in der Per-Protocol(PP)-Analyse (n = 21) sind in der untenstehende Tabelle 3 zusammengefasst.
Zusätzlich wurde die inkrementelle Wiederfindungsrate (In Vivo Recovery [IVR]) aus Spiegeln bis zu 4 Stunden nach der Infusion bestimmt. Die mediane inkrementelle IVR war ein Anstieg um 1,8 mg/dL (Bereich 1,08 – 2,62 mg/dL) pro mg/kg. Die mediane IVR besagt, dass eine Dosis von 70 mg/kg die Fibrinogenkonzentration im Plasma des Patienten um ca. 125 mg/dL erhöht.
Tabelle 3: Pharmakokinetische Parameter (n = 21) für die Fibrinogenaktivität
(PP-Population*)
| Parameter | Mittelwert ± SD | Bereich |
|---|---|---|
| Halbwertszeit (h) | 75,9 ± 23,8 | 40,0 – 157,0 |
| Cmax (mg/dL) | 139,0 ± 36,9 | 83,0 – 216,0 |
| AUCnorm für Dosis von 70 mg/kg (mg*h/mL) | 113,7 ± 31,5 | 59,7 – 175,5 |
| Clearance (mL/h/kg) | 0,67 ± 0,2 | 0,4 – 1,2 |
| Mittlere Verweildauer (h) | 106,3 ± 30,9 | 58,7 – 205,5 |
| Verteilungsvolumen im Steady State (mL/kg) | 70,2 ± 29,9 | 36,9 – 149,1 |
* Ein Patient wurde von der PP-Population ausgeschlossen, weil dieser < 90 % der vorgesehenen
Dosis von Fibryga und dem Vergleichspräparat erhalten hatte
Cmax = maximale Plasmakonzentration; AUCnorm = für die verabreichte Dosis normalisierte
Fläche unter der Kurve; SD = Standardabweichung
Pharmakokinetik bei besonderen Patientengruppen
Zwischen männlichen und weiblichen Studienteilnehmern wurde hinsichtlich der Fibrinogenaktivität kein statistisch relevanter Unterschied festgestellt. In der PP-Analyse wurde bei der Halbwertszeit bei Patienten unter 18 Jahren (n = 5) mit 72,8 ± 16,5 Stunden ein geringer Unterschied gegenüber der Gruppe der Erwachsenen (n = 16, 76,9 ± 26,1 Stunden) beobachtet. Die Clearance war in beiden Altersgruppen nahezu identisch und betrug 0,68 ± 0,18 mL/h/kg bzw. 0,66 ± 0,21 mL/h/kg.
Kinder und Jugendliche
Für Kinder unter 12 Jahren liegen keine pharmakokinetischen Daten vor.
5.3 Präklinische Daten zur Sicherheit
Die Sicherheit von Fibryga wurde in mehreren präklinischen Studien zur Sicherheitspharmakologie (kardiovaskuläre Wirkungen, thrombogenes Potential) und zur Toxikologie (akute Toxizität, lokale Verträglichkeit) untersucht. Basierend auf diesen Studien lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen. Im Venostase-Test (Wessler- Test) war Fibryga bei Dosen von bis zu 400 mg/kg Körpergewicht nicht thrombogen.
6. Pharmazeutische Angaben
6.1 Liste der sonstigen Bestandteile
Pulver
- L-Argininhydrochlorid
- Glycin
- Natriumchlorid
- Natriumcitrat-Dihydrat
Lösungsmittel
- Wasser für Injektionszwecke
6.2 Inkompatibilitäten
Das Arzneimittel darf nicht mit anderen Arzneimitteln gemischt werden.
6.3 Dauer der Haltbarkeit
2 Jahre.
Die chemische und physikalische Stabilität nach Rekonstitution wurde für 24 Stunden bei Raumtemperatur (max. 25 °C) gezeigt. Aus mikrobiologischer Sicht sollte das Arzneimittel sofort nach der Rekonstitution verwendet werden. Wenn die rekonstituierte Lösung nicht sofort angewendet wird, unterliegen die Aufbewahrungszeiten und -bedingungen der Verantwortung des Anwenders. Die rekonstituierte Lösung darf nicht eingefroren oder im Kühlschrank gelagert werden. Angebrochene Flaschen sind zu verwerfen.
6.4 Besondere Vorsichtsmaßnahmen
für die Aufbewahrung Nicht über 25 °C lagern. Nicht einfrieren. Die Flasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen. Aufbewahrungsbedingungen nach Rekonstitution des Arzneimittels, siehe Abschnitt 6.3.
6.5 Art und Inhalt des Behältnisses
Jede Packung enthält:
- 1 g humanes Fibrinogen in einer farblosen 100-mL-Glasflasche, Typ II Ph. Eur., verschlossen mit einem Infusionsstopfen (Brombutylkautschuk) und einer Verschlusskappe aus Aluminium
- 50 mL Lösungsmittel (Wasser für Injektionszwecke) in einer farblosen 50-mLGlasflasche, Typ II Ph. Eur., verschlossen mit einem Infusionsstopfen (Halobutylkautschuk) und einer Verschlusskappe aus Aluminium
- 1 Octajet-Transfersystem
- 1 Partikelfilter
6.6 Besondere Vorsichtsmaßnahmen für die Beseitigung und sonstige Hinweise zur Handhabung
Allgemeine Hinweise
- Die rekonstituierte Lösung sollte nahezu farblos und leicht opaleszierend sein.
- Keine Lösungen verwenden, die trübe sind oder Ablagerungen aufweisen.
Rekonstitution
- Das Pulver (Fibryga) und das Lösungsmittel (WFI) in ungeöffneten Flaschen auf Raumtemperatur bringen. Diese Temperatur sollte während der Rekonstitution beibehalten werden. Wenn zum Aufwärmen ein Wasserbad benutzt wird, muss darauf geachtet werden, dass das Wasser nicht in Kontakt mit den Gummistopfen oder den Verschlusskappen der Flaschen kommt. Die Temperatur des Wasserbads sollte + 37 °C nicht überschreiten.
- Verschlusskappen von den Flaschen mit dem Pulver (Fibryga) und dem Lösungsmittel abziehen, um den zentralen Teil des Infusionsstopfens freizulegen. Die Gummistopfen der beiden Flaschen mit einem Alkoholtupfer säubern und anschließend trocknen lassen.
- Deckel von der äußeren Verpackung des Octajet-Transfersystems abziehen. Das Octajet-System in der durchsichtigen äußeren Verpackung belassen, um die Sterilität zu gewährleisten.
- Das Octajet-Transfersystem mit der äußeren Verpackung aufnehmen und über der Flasche mit dem Pulver (Fibryga) umdrehen. Das in der äußeren Verpackung befindliche Transfersystem in der Mitte der Flasche mit dem Pulver aufsetzen, bis die Klemmen des (farblosen) Einstechdorns für das Arzneimittel eingerastet sind. Die Pulverflasche festhalten und vorsichtig die äußere Verpackung vom Octajet entfernen, wobei der (blaue) Dorn für das Wasser nicht berührt werden darf und das Octajet fest mit der Konzentratflasche verbunden bleibt.
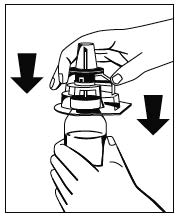
Abb. 1 - Während die Flasche mit dem Pulver (Fibryga) auf einer ebenen Oberfläche festgehalten wird, die Flasche mit dem Lösungsmittel umdrehen und in der Mitte des Einstechdorns für die Wasserflasche platzieren. Den blauen Kunststoffdorn des Octajets fest durch den Gummistopfen der Flasche mit dem Lösungsmittel drücken.
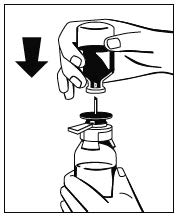
Abb. 2 - Den Abstandhalter entfernen (Abb. 3) und die Flasche mit dem Lösungsmittel herunterdrücken (Abb. 4). Darauf fließt das Lösungsmittel in die Flasche mit dem Pulver (Fibryga).
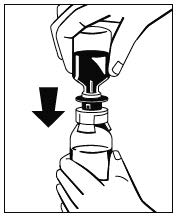
Abb. 4 - Wenn die Überführung des Lösungsmittels abgeschlossen ist, die Flasche mit dem Arzneimittel vorsichtig schwenken, bis das Pulver vollständig gelöst ist. Die Flasche nicht schütteln, um Schaumbildung zu vermeiden. Das Pulver sollte innerhalb von ca. 5 Minuten vollständig gelöst sein. Das Auflösen des Pulvers sollte nicht länger als 30 Minuten dauern. Wenn das Pulver nach 30 Minuten noch nicht vollständig aufgelöst ist, muss das Arzneimittel verworfen werden.
- Den blauen Anschluss der Lösungsmittelflasche drehen (in beide Richtungen möglich), um die Markierungen aneinander auszurichten, und dann die Lösungsmittelflasche zusammen mit dem Wasserdorn entfernen. (Abb. 5)
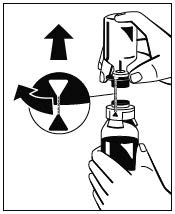
Abb. 5 - Eine Spritze am beiliegenden Filter anbringen (Abb. 6) und den Filter am Luer-Lock-Anschluss des Octajets auf der Pulverflasche anschließen (Abb. 7). Lösung durch den Filter in die Spritze ziehen. (Abb. 8)
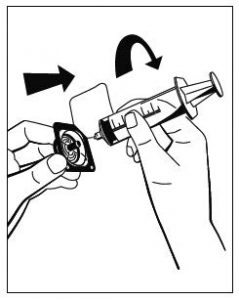
Abb. 6 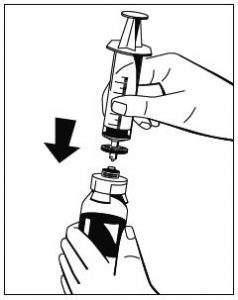
Abb. 7 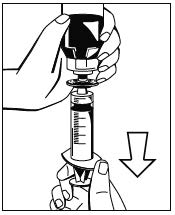
Abb. 8 - Die gefüllte Spritze vom Filter abnehmen und die leere Flasche verwerfen. Zur intravenösen Verabreichung der rekonstituierten Lösung bei Raumtemperatur wird die Verwendung eines herkömmlichen Infusionssets empfohlen. Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
7. Inhaber der Zulassung
OCTAPHARMA GmbH
Elisabeth-Selbert-Str. 11
40764 Langenfeld
E-Mail: info.de@octapharma.com
www.octapharma.de
8. Zulassungsnummer(n)
PEI.H.11879.01.1
9. Datum der Erteilung der Zulassung/Verlängerung der Zulassung
Datum der Erteilung der Zulassung: 23. Juni 2017
10. Stand der Information
10.2019
11. Verkaufsabgrenzung
Verschreibungspflichtig.
12. Herkunftsland des Blutplasmas
Deutschland, Estland, Kroatien, Lettland, Litauen, Luxemburg, Norwegen, Österreich, Polen, Portugal, Schweden, Schweiz, Slowenien, Tschechische Republik, Ungarn, USA